|
|
| Overcoming the Limitations of Directed C–H Functionalizations of Heterocycles |
 |
Text Size: A A A |
|
Heterocycles are commonly found in drug candidates, and in medicinal chemistry research. It is well appreciated that the presence of a heterocycle can improve solubility and reduce the lipophilicity of a drug molecule. The potential application of C–H activation technologies in the rapid synthesis and diversification of novel heterocycles has attracted widespread attention from the pharmaceutical industry. One of the most significant challenges in the application of C–H functionalization reactions is achieving robust control of positional selectivity. Directed C–H metalation has recently emerged as a reliable approach for achieving a diverse collection of selective C–H functionalization reactions, and activation of both proximate and remote C–H bonds have proven feasible. The use of a weakly coordinating functional group to achieve high effective molarity of the catalyst around the C–H bond of interest has greatly expanded the substrate scope of these processes. Unfortunately, these C–H functionalization processes are generally incompatible with the majority of medicinally important heterocyclic substrates because the heteroatoms can interfere with the catalyst. Strongly coordinating nitrogen, sulfur, and phosphorous heteroatoms outcompete the directing groups for catalyst binding, thus preventing activation of the C–H bonds proximate to the directing groups. Professor Jin-quan Yu research group from State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences recently in Nature published an article titled "Overcoming the Limitations of Directed C–H Functionalizations of Heterocycles" article (Nature2014, DOI: 10.1038/nature13885). They report a robust and synthetically useful reaction that overcomes the complications associated with performing C–H functionalization reactions on heterocycles. Their approach employs a simple N-methoxy amide group, which serves as both a directing group and an anionic ligand to promote the in situ generation of the reactive PdX2 (X = ArCONOMe) species from precursor from a Pd(0) source using air as the sole oxidant. In this way, the PdX2 species is inherently anchored in close proximity with the target C–H bond adjacent to CONHOM e group, thus avoiding the interference from various heterocycles. The C–H functionalization products can be readily converted to versatile synthons containing a variety of functional groups. Remarkably, this reaction overrides the conventional positional selectivity patterns observed with substrates containing strongly coordinating heteroatoms, including nitrogen, sulfur, and phosphorus. Thus, this operationally simple aerobic reaction demonstrates the feasibility of bypassing a fundamental limitation that has long plagued applications of directed C–H activation in medicinal chemistry. 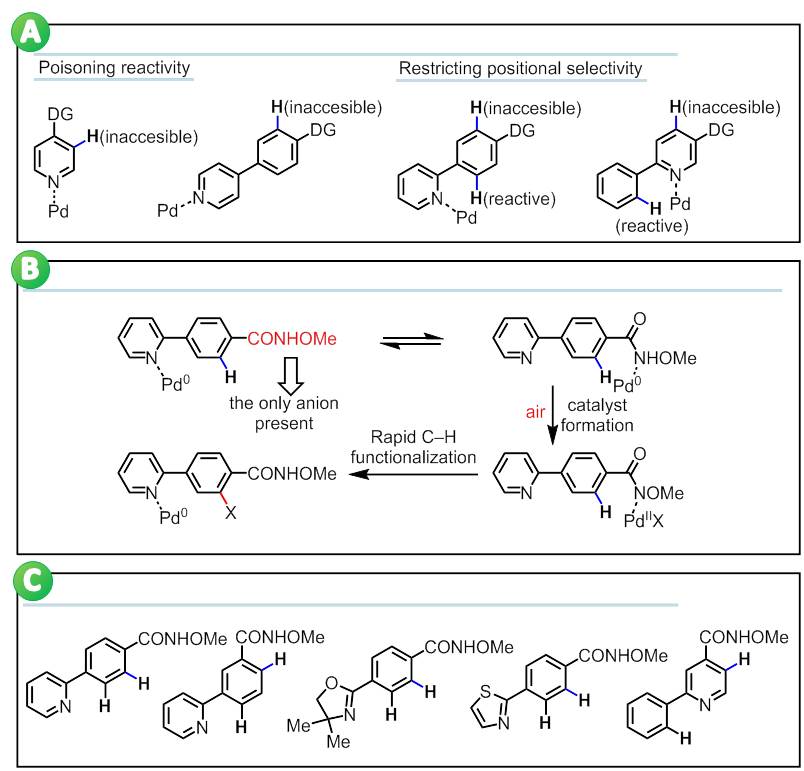
A) Fundamental limitation of directed C-H functionalization of heterocycles; B) Approach: on-site generation of a Pd(II) catalyst assisted by the anionic directing group; C) Overiding site-selectivity dictated by the strongly coordinating heterocycles.(Imaged by YU Jin-quan@SIOC) |
|
|
