Propargylic substitution has been extensively studied as an efficient way to prepare chiral alkyne or allene skeletons. In this case, a heteroatom leaving group should be pre-stored at the vicinal position of alkyne unit. If the leaving group lies remote to the alkyne units through an inert alkyl chain, the substitution process is considered unfeasible. In a recent work published in Science Advances, a research team led by Prof. HE Zhi-Tao at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences designed a palladium/amine synergistic catalysis strategy for the asymmetric remote substitution of alkynes (Figure 1). Mechanistic experiments revealed a complex redox change at palladium metal center and the formation of conjugated diene intermediate. In 2023, HE and coworkers first uncovered that internal alkyne could underwent the formation of conjugated diene prior to hydrofunctionalization. Inspired by this mechanistic discovery, they made a rational substrate design by introducing a leaving group at the γ position of alkyne substrates. They hypothesized that if the conjugated diene was generated quickly from alkyne prior to hydrofunctionalization, the remote leaving group at the γ position might be cleaved through the oxidation addition of low-valent metal by the newly formed diene intermediate. Researchers used 2-phenylpropanal as the nucleophile, diglyme as the solvent, and PhCO2H as the additive to realize the design, which in turn proved the hypothesis feasible. A set of newly modified diamine organocatalysts were critical to guarantee the excellent regio-, geometry- and enantioselectivity of the transformation. The scope was broad and a series of aryl and ester-derived alkynes were compatible with the reaction. Scale-up test was efficient and a series of downstream derivatizations were carried out to prepare various enantioenriched skeletons. Mechanistic experiments were then conducted to unveil the possible pathways of the reaction. Catalyzed by PdH species, a crucial conjugated diene intermediate was generated from alkyne by β-H elimination. Then the PdH catalyst underwent reductive elimination to give Pd(0) species, which could catalyze an asymmetric allylation substitution to obtain the product from the diene intermediate. Deuterium labelled experiments suggested that the hydrofunctionalization of diene, allene or alkyne was strictly inhibited during the transformation. Kinetic experiments and kinetic isotope effect illustrated that the rate-determining step might be the alkyne isomerization. In summary, a novel asymmetric remote substitution of alkynes via Pd/chiral amine synergetic catalysis is established in excellent yield and selectivity. Mechanism experiments support the formation of diene intermediate from alkyne and a complex redox pathway of Pd catalyst. 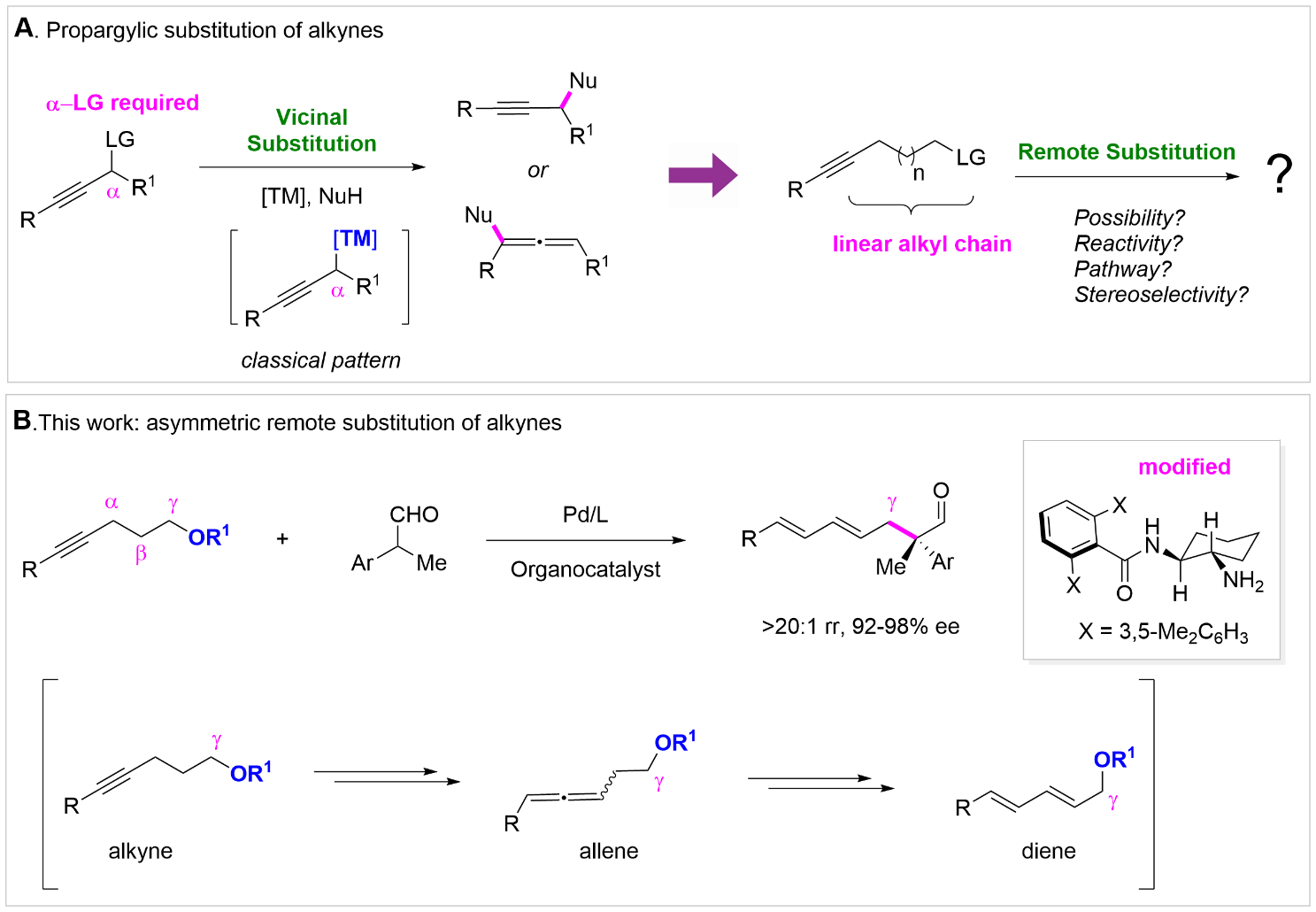
Figure 1. Asymmetric remote substitution of alkynes (Image by HE ZhiTao)
Prof. HE Zhi-Tao Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences 345Lingling Rd. Shanghai, 200032, China. Tel: 86-21-54925081 E-mail: hezt@sioc.ac.cn |
