Dearomatization reactions represent a cornerstone strategy for the construction of pharmacologically important saturated cyclic scaffolds. Photoexcitation modulates the electron distribution in the frontier molecular orbitals of aromatic compounds, enabling dearomatization pathways that are inaccessible under ground-state conditions and delivering structurally unique molecular architectures. In a recent invited review published in Science Advances, a research team led by Prof. YOU Shu-Li at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, introduced the excited-state dearomatization and highlighted representative examples based on three distinct excitation modes: direct excitation, energy-transfer (EnT) photocatalysis, and exciplex formation (Figure 1). The first mode involves direct excitation. To illustrate this pathway, the work by the Leonori group was highlighted, in which direct excitation of nitroarenes was employed to achieve their photochemical conversion into bicyclic pyrrolines. Peripheral coordination of aromatic substrates with a Lewis acid catalyst or promoter gives rise to a pronounced bathochromic shift, thereby enabling facile and selective excitation of the resulting complexes. This strategy suppresses racemic background reactions and permits efficient control over the enantioselectivity in dearomatization processes. This research field was pioneered by the groups of Meggers, Glorius, Bach, YOU, and Dell'Amico. The second mode refers to energy-transfer photocatalysis. This activation strategy was pioneered by the Glorius group for the dearomatization of 1-naphthol derivatives with alkenes. The You group demonstrated the viability of EnT-triggered dearomatization of indole derivatives through intramolecular [2+2] photocycloaddition. For azaarene substrates with high triplet energies, external additives are often necessary to render them susceptible to photosensitizer-mediated excitation. In this context, Brown, Houk, Glorius, and co-workers employed BF₃·OEt₂ or hexafluoroisopropanol (HFIP) as acidic additives to lower the triplet energies of (iso)quinolines, thereby facilitating their dearomative [4+2] photocycloaddition with alkenes. Beyond photocycloaddition manifolds, highly reactive triplet-state aromatics participate in diverse dearomative transformations. The König group realized Birch-type photoreduction of (hetero)arenes under photocatalytic conditions. The Bach group developed a distinct class of transformations involving photocatalytic dearomative hydrogen-atom transfer (HAT)/cyclization cascades of indole derivatives. The direct excitation and EnT strategies described above necessitate the modification of substrates with auxochromes or directing groups; in the absence of such moieties, exciting these substrates to their triplet excited states remains highly challenging. Exciplex formation provides an effective strategy to circumvent this limitation. A notable example was reported by the Sarlah group, which introduced visible-light-photoactivable 2π components as "arenophiles" to enable exciplex-mediated dearomatization. Additionally, the Leonori group described a visible-light-driven ozonolytic deconstruction of aromatics, mediated by exciplex formation between excited nitroarenes and electron-rich arenes. This approach addresses the over-oxidation issue inherent to classical aromatic ozonolysis, a long-standing challenge in this field. Several avenues regarding future research directions have been proposed. Although dearomative cycloadditions have attracted considerable attention, non-cycloaddition-based dearomatization reactions, aimed at expanding the diversity of saturated cyclic scaffolds, deserve greater research efforts. The exploration of enantioselective control in excited-state dearomatization reactions remains relatively limited, representing a key area for further investigation. Notably, the rational design of efficient synthetic strategies that favor kinetically less reactive dearomatization processes while suppressing the undesired overreaction of the resulting products has emerged as an imperative research objective. Furthermore, advanced characterization techniques and computational modeling approaches are essential to unravel the underlying reaction mechanisms of excited-state dearomatization. In summary, photon energy can be harnessed to disrupt aromaticity, rendering dearomatization reactions thermodynamically and kinetically favorable under mild conditions. A wide range of aromatic substrates, including naphthalenes and benzenes, have been converted into (partially) saturated cyclic scaffolds through such strategies. This invited review highlights representative visible-light-driven excited-state dearomatization reactions, encompassing the three distinct excitation modalities discussed above, and also proposes several avenues for future research in this field. 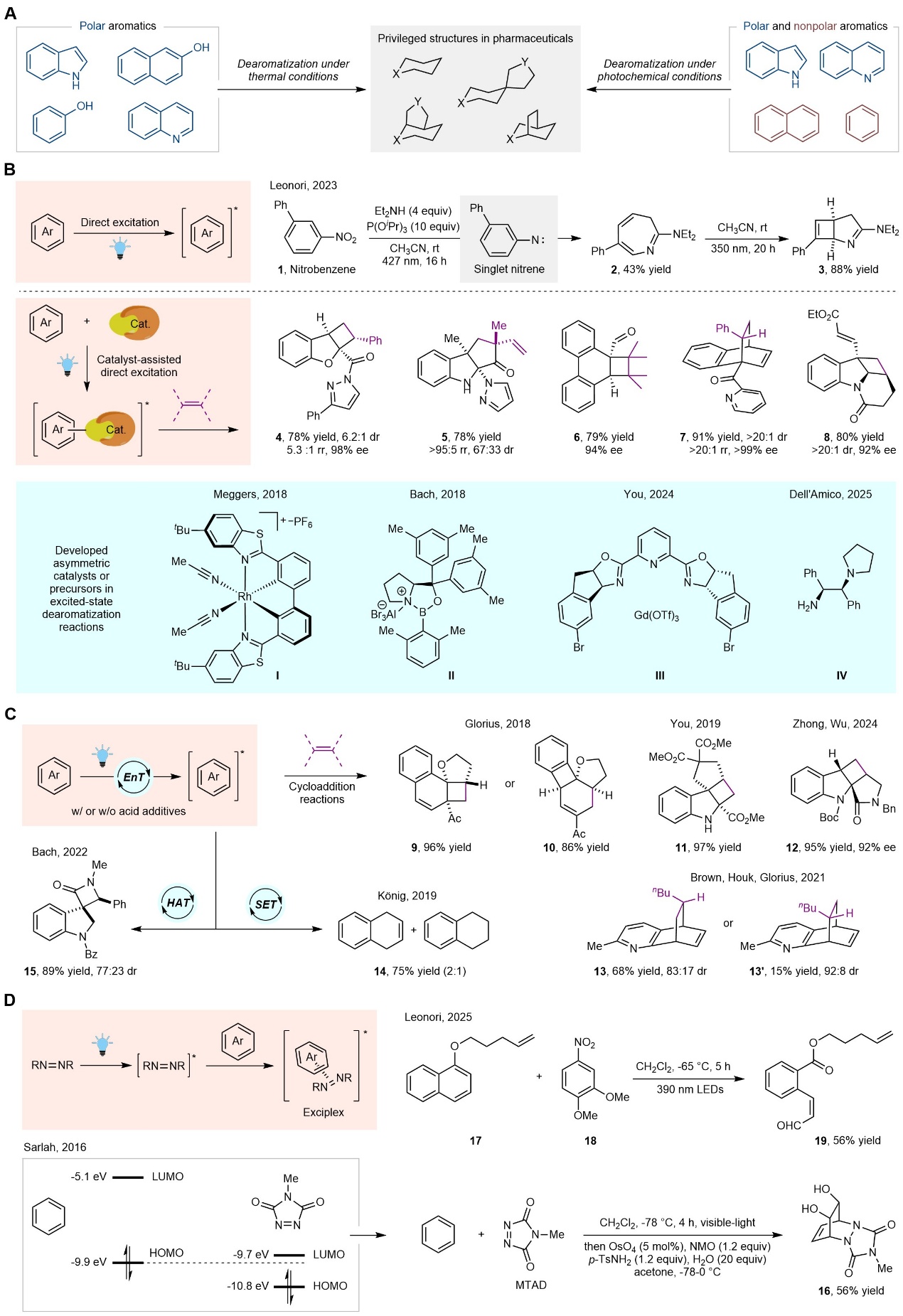
Figure 1.Visible-light-enabled excited-state dearomatization reactions (Image by YOU Shu-Li)
YOU Shu-Li Ph.D. Professor Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences Ling Ling Road 345 Shanghai 200032 China Tel: 0086-21-54925085 Email: slyou@sioc.ac.cn |
